Co-Cu合金の表面モデリング
ここではCo-Cu合金の表面モデリングを行う方法を説明します。合金材料を利用した表面反応は合金表面における組成や原子位置に依存して性能が変化します。そのため、合金の表面状態を知ることは表面反応の理解に向けて重要であるとされています。
そこで、量子アニーリングを利用したCo-Cu合金の表面モデリング方法を本項で説明します。
以下に全体の流れの概略図を示します。
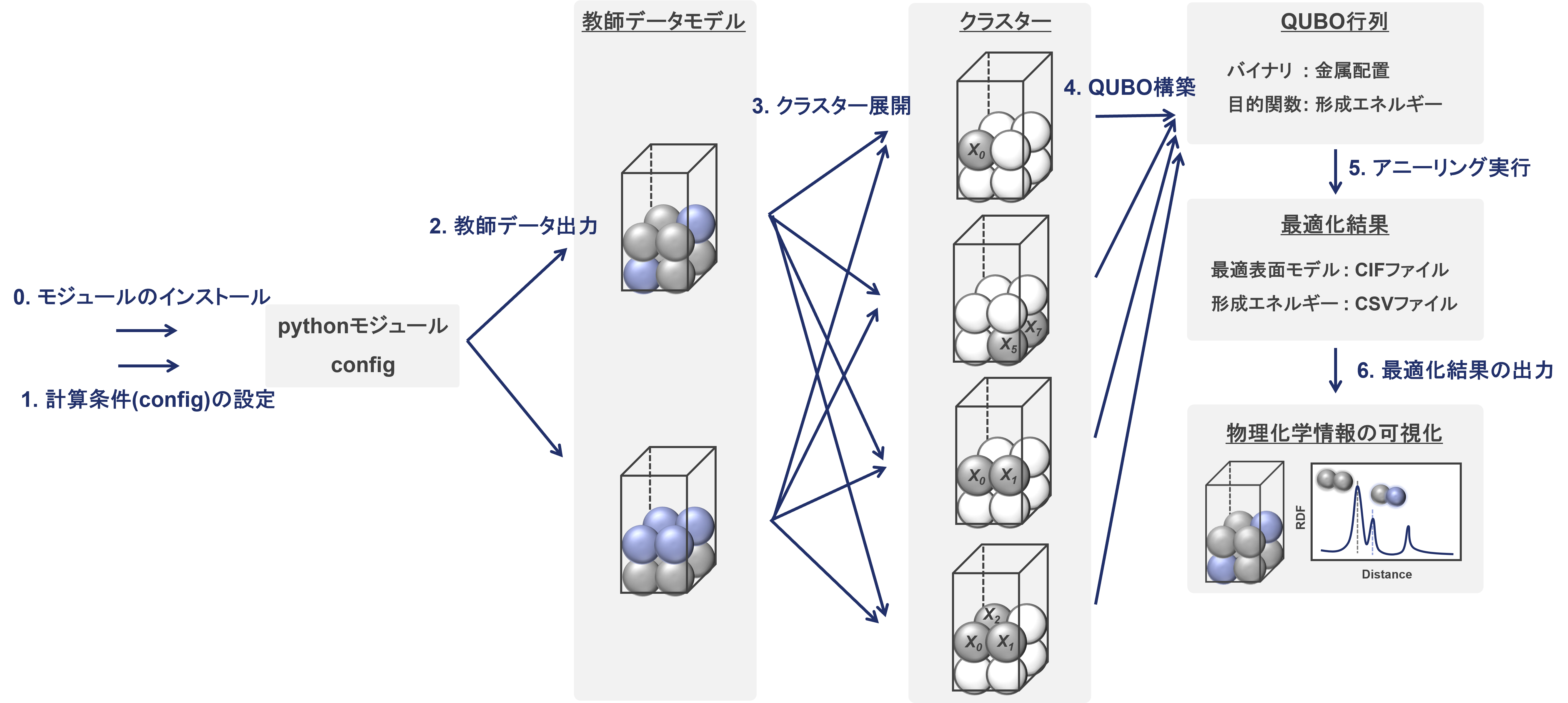
0. 必要なモジュールのインストール (初回のみ実行)
実行環境によって必要なモジュールがない可能性があるため、初回実行時に必要なモジュールをインストールしてください。
1pip install ase==3.22
2pip install spglib
3pip install cycthon
4pip install clease
5pip install tqdm
6pip install amplify
7pip --no-cache-dir install amplify_bbopt
8sudo apt-get install python3.11-dev
9python cython_setup.py build_ext --inplace
10cythonize -i -a cython_process.pyx
1. configファイルによる条件設定
本ソフトウェアはconfigファイルに記載された実行条件を読み込んで実行されます。
デフォルトの計算条件は既にconfigファイルに記載されているため、まず初めにデフォルトのconfigファイルを読み込み、必要に応じて計算条件を変更します。
1import allfunc
2config = allfunc.Prepare_Config()
3config.read()

configファイルは以下のSectionで構成されています。
preparation:求めたい合金表面の情報に関する設定
cluster:クラスター展開に関する設定
calculation:教師データに関する設定
annealing:アニーリングの実行に関する設定
対象とする材料系や計算条件をここで設定します。
ここでは以下の条件をそれぞれ「config.write("Section name","key name", "value")」のフォーマットで指定します。
storage_path (実行場所の指定)
substitute_ratio (扱う合金種)
supercell_multiple (扱う表面モデルサイズ)
host_path (元となるバルク構造のCIFファイルのパス)
このほかにも、検討する露出面や表面モデルの真空層についてもconfigで設定することができます。
1#Exection location
2config.write("preparation","storage_path","./work/")
3allfunc.storage_path_initialize(config)
4
5#Target alloy composition
6config.write("preparation","substitute_ratio",[{"Cu": [0.0, 1.0], "Co": [0.0, 1.0]}])
7
8#Surface model size
9config.write("preparation","supercell_multiple",[3,3,3])
10
11#Path to the CIF file of the initial bulk structure
12config.write("preparation","host_path",'./bulk.cif')
13
14#For check
15config.read()
2. 教師データ用モデルの出力とエネルギー取得
次に、金属配置問題の定式化に向けたクラスター展開法を行うための教師データを作成します。
教師データの作成に向けて、以下の条件を設定する必要があります。
train_data_num (教師データの数)
様々な金属比率や金属配置を持つ表面モデルに対する形成エネルギーの関係性が多くあると、金属配置問題を正確に定式化することに繋がるため、教師データ数は多い方が良いです。
しかし、対象とする表面モデルのサイズで構成可能な非等価な表面モデルは最大2^N通りで、表面対称性を考慮するとさらに非等価な表面モデルは限られてくるため、ここでの教師データの数はその値より小さくする必要があります。
calculation_method (教師データを算出する方法)
VASPで算出する場合:'["VASP","VASP"]'
QuantumEspressoで算出する場合:'["QuantumEspresso","QuantumEspresso"]'
エネルギー値をCSVから読み込む場合:'["csv_reader","csv_read"]'
fmax (教師データ算出時の収束閾値)
max_cluster_size (クラスター展開でのクラスターサイズ)
検討するクラスター原子数ごとのクラスターサイズをリスト形式で指定します。
クラスターサイズは検討する表面モデルサイズより小さい必要があります。
また、このリストの長さが検討するクラスター原子数を指定するため、[3.0]と指定した場合は3Åの2原子クラスターを考慮し、[3.0, 5.0]と指定した場合は3Åの2原子クラスターと5Åの3原子クラスターを考慮した定式化を行います。
1#Number of training data
2config.write("calculation","train_data_num",'30')
3
4#Method for calculating the training data
5config.write("calculation","calculation_method",'["csv_reader","csv_read"]')
6
7#threshold of convergence on the training data calculation
8config.write("calculation","fmax",'0.02')
9
10#Cluster size on cluster expansion
11config.write("cluster","max_cluster_size", '[3.5]')
計算条件を設定できたら、以下の関数により教師データ用のデータセットCSVと表面モデルを出力します。
1CE = allfunc.ClusterExpansionExcutor(config)
2CE.model_addition_random()
ここで得られた教師データ用表面モデルを用いて形成エネルギーを算出します。
以下のプログラムにおいて、configファイルのcalculation_methodで設定した方法により形成エネルギーを算出または読み込みすることで、教師データ用のデータセットCSVに形成エネルギーを記載します。
1CE.unconverged_calculate()
3. クラスター展開によるQUBO係数の算出
次に、アニーリングマシンでの求解に向けてQUBO係数をクラスター展開により算出します。
ここで以下の条件によりQUBO係数の回帰結果が左右されるため、必要に応じて条件設定を行ってからクラスター展開を行います。
scoring_scheme (回帰に向けた評価方法)
デフォルトではk-分割交差検証が採用されています。他の評価手法を利用する場合は条件を変更してください。
split_number (交差検証における分割数)
fitting_scheme (回帰スキーム)
alpha (回帰における係数)
1CE.ECI_evaluate()
このときに、実行条件下での精度評価が行われます。
ここではクロスバリデーションによる評価を行っており、規定値よりも大きい値の場合はクラスター展開における条件を変えることで精度をあげることが可能となります。
4. QUBO行列の構築
クラスター展開により回帰されたQUBO係数を用いてQUBO行列を構築します。
求解を行うアニーリングマシンの種類によって構築するQUBO行列が異なるため、ここでアニーリングマシンの設定を行う必要があります。
今回はFixstars Amplifyのアニーリングマシンを使用することとします。
1QUBO,constant=CE.make_QUBO()
得られたQUBO行列を確認すると、指定した次数に対応した行列のshapeになっていることが分かり、各係数を足し合わせることである金属配置での形成エネルギーを導出することができます。
5. アニーリングの実行
次に、クラスター展開により構築したQUBO行列を読み込み、アニーリングマシンで表面エネルギーが最小となる金属配置を求めていきます。
目的の組成があればその組成だけに注目した実行も可能ですが、今回はCuに対してCoを3原子ずつ置換した際の表面構造を探索するプログラムを示します。
1from ase.io import write
2import os
3import pandas as pd
4
5#Annealing setting
6fa = allfunc.Fixstars_Amplify(config,QUBO,constant,CE.make_qubit())
7fa.set_default_penalty(QUBO,"QUBO_max")
8fa.default_penalty *= 10
9
10#Result path setting
11resultdir='./work/result/'
12if not os.path.isdir(resultdir):
13 os.makedirs(resultdir)
14columns=["Cu","Co","energy"]
15df = pd.DataFrame(columns = columns)
16
17#Execute annealing
18for i in range(0,28,3):
19 result = fa.run([None,None],{"Cu":i,"Co":27-i})
20 result_dict = fa.decode_to_universal_style(result)
21 write(resultdir+f"Cu{i}Co{27-i}.cif",allfunc.best_bits2model(result_dict,CE))
22 df = df._append(pd.Series([i,27-i,result_dict["best_energy"]],index=df.columns,name=str(i)))
23df.to_csv(resultdir+"energy.csv")
result_dictというdictには、検討している組成比における最適な金属配置情報を表示することができます。
best_energy: 最適な金属配置における形成エネルギー
best_bits: 最適な金属配置におけるバイナリ (これをもとにCIFファイルを構築しています。)
6. 最適化結果の出力
アニーリングの実行が終了すると、最適化後の表面モデルのCIFファイルとその形成エネルギーのCSVファイルを得ることができます。
この出力だけでも十分ですが、最適化後の表面モデルに対して以下に示す物理化学的情報を可視化することができ、合金構造の特徴を確認することができます。
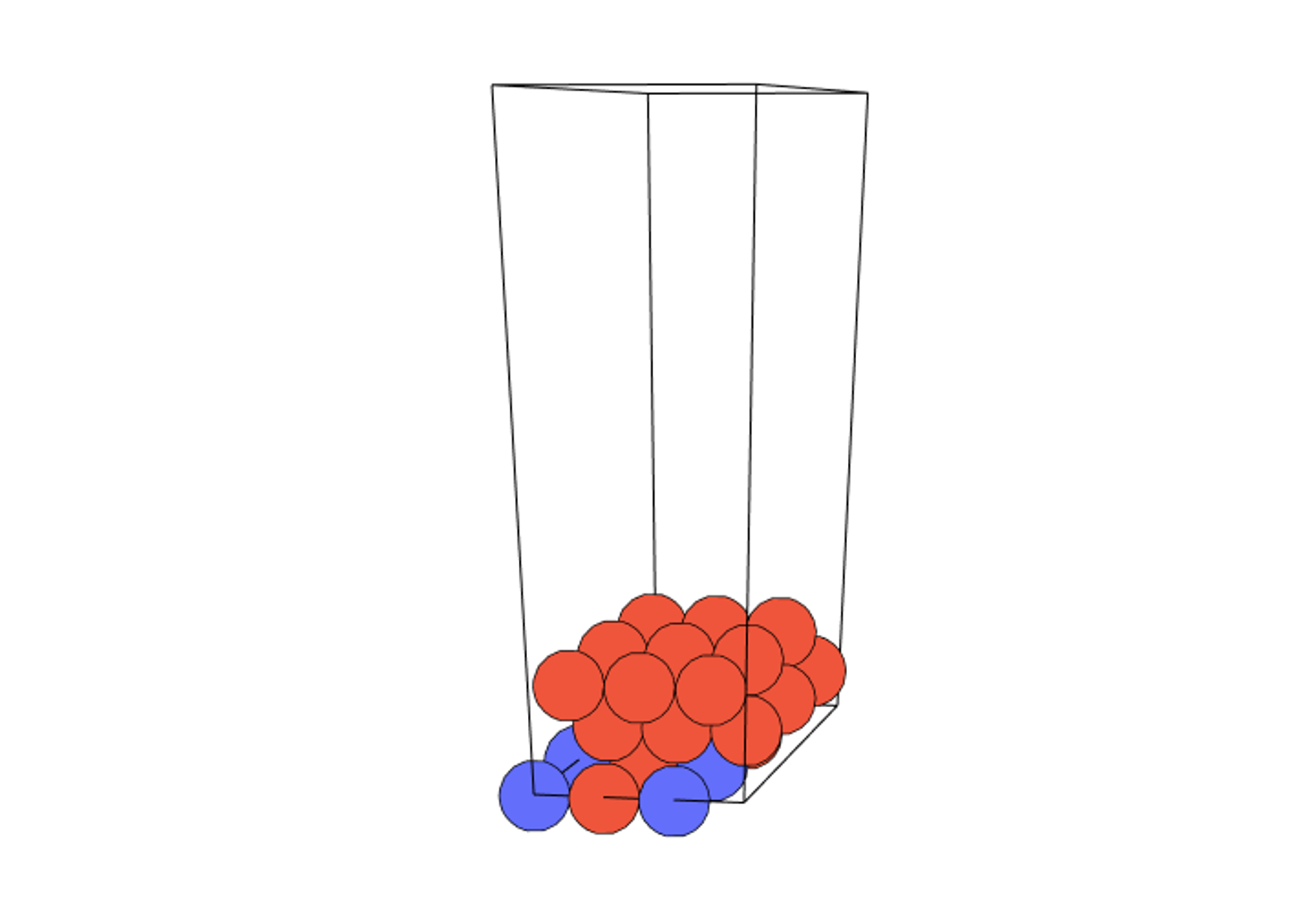
表面モデルにおける金属位置の描画
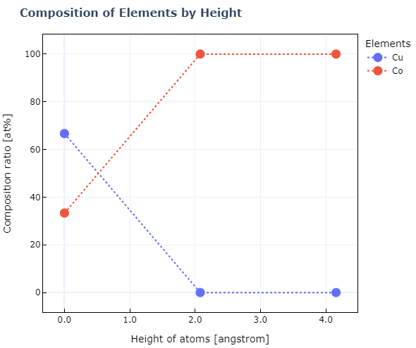
表面モデルの深さ方向の組成分布
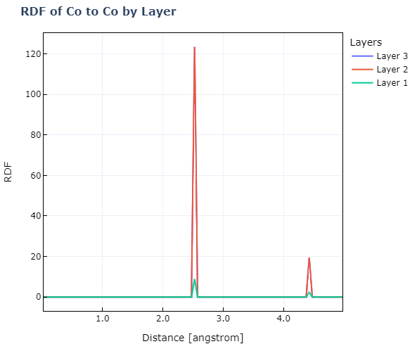
表面モデルの層ごとの動径分布関数
各組成比に対する2元素間の相対高さ
各組成比に対する形成エネルギー
1import visualize
2elements = ['Cu','Co']
3total_atoms = 27
4resultdir = './work/result/'
5
6visualization_manager = visualize.VisualizationManager(resultdir, elements, total_atoms)
1#Visualization of metal positions in the surface model
2alloy_ratios=[3,24]
3visualization_manager.view_model(alloy_ratios,size=22)
1#Depth-wise composition distribution of the surface model
2alloy_ratios=[6,21]
3visualization_manager.view_composition(alloy_ratios)
1#Radial distribution function for each layer of the surface model
2alloy_ratios=[6,21]
3visualization_manager.view_rdf(alloy_ratios, center_element='Co', target_elements=['Co'])
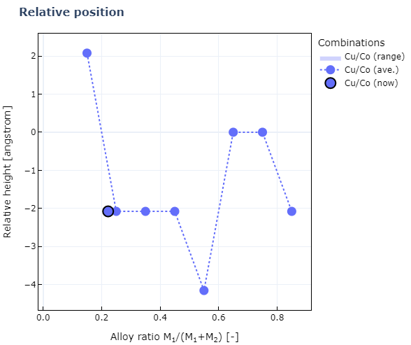
1#Relative heights between two elements for each composition ratio
2alloy_ratios=[6,21]
3visualization_manager.view_relativeposition(alloy_ratios)
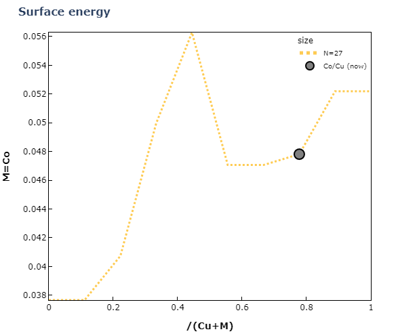
1#Formation energy for each composition ratio
2alloy_ratios=[6,21]
3visualization_manager.view_energy(alloy_ratios, visualize_start=0, visualize_width=3)
また、組成比を変えた複数の表面モデルの探索を行った場合、組成比と表面構造の関係性をインタラクティブに理解するためのダッシュボードを利用することが可能です。
1import visualize
2import dashboard
3
4elements = ['Cu','Co']
5total_atoms = 27
6resultdir = './work/result/'
7
8visualization_manager = visualize.VisualizationManager(resultdir, elements, total_atoms) # VisualizationManagerのインスタンスを生成
9app = dashboard.DashApp(elements, total_atoms, visualization_manager, initial_plots=['model','composition', 'rdf','relativeposition','energy']) # 初期プロットを設定
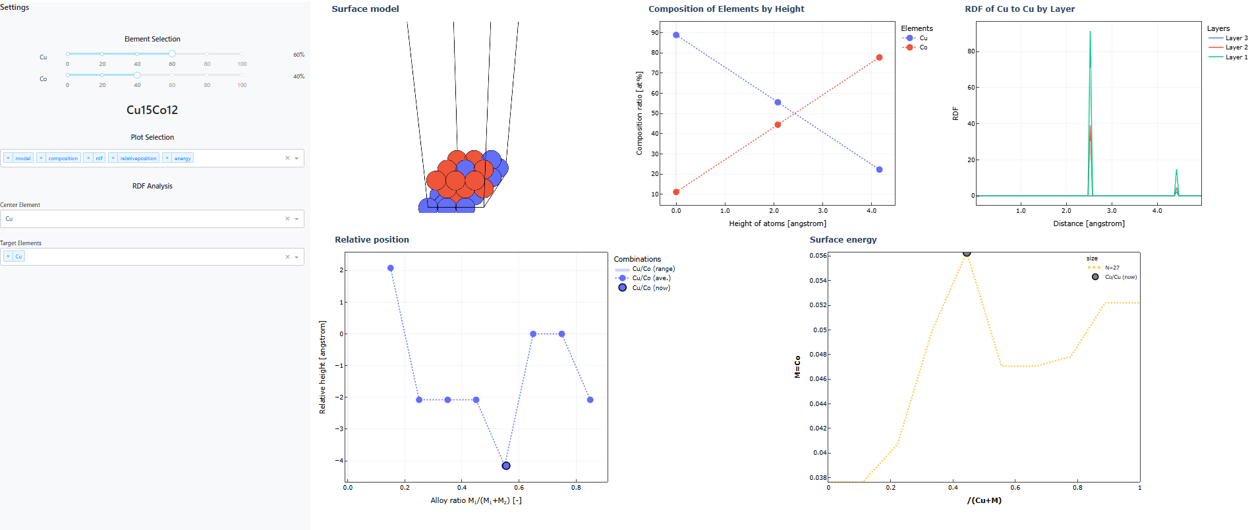
上のコードで出力されたダッシュボードにおいて、左のsliderを動かすと様々な組成比における表面モデルの物理化学的情報をまとめて描画することが可能です。
この結果から、Cu-Co合金では規則的に混ざるよりも相分離したほうが安定であることが分かります。
ただし、クラスター展開による定式化の精度が未だ低いことなどから、結果が妥当かどうかは別で検討する必要があります。
また、このソフトウェアにおいて計算条件や検討する系を変えることで、様々な系に対する金属配置について様々な情報を得ることができます。
7. 最適化モデルの利用 (オプション)
最適な金属配置を持った合金表面モデルは、触媒反応や表面反応の理解に向けて様々な原子や分子の吸着挙動を評価することになります。
このソフトウェアでは表面の対称性を取得して、非等価な吸着モデルを出力することができます。
現状H原子やCO分子といった単純な系に限られていますが、今後CO2分子やH2Oの吸着にも対応できるように開発を進めています。